Chemical Heuristics in the 21st Century
Chemical Heuristics in the 21st Century
Predicting chemical reactions is hard. Without experimental data or prior experience, the enterprising theoretical chemist is forced to examine the potential energy surface (PES) of the reaction, tracing the energy valleys from the reactants to potential products in a high-dimensional space, searching for favorable transition states—the mountain passes on the energy landscape—relevant to chemical transformations. The brute-force solution of searching the entire PES is a computationally prohibitive task, which is why many sophisticated techniques of PES exploration have been developed.1
Alternatively, we can pick any first-semester textbook of organic chemistry to learn how to predict reactions without worrying about any of the complexities of the PES. The first-semester textbook will teach us heuristics—simple rules for finding plausible chemical reactions, however, without guaranteed success. Heuristics are a time-tested approach for solving computationally hard tasks, for example, the A* search algorithm in robotics.2
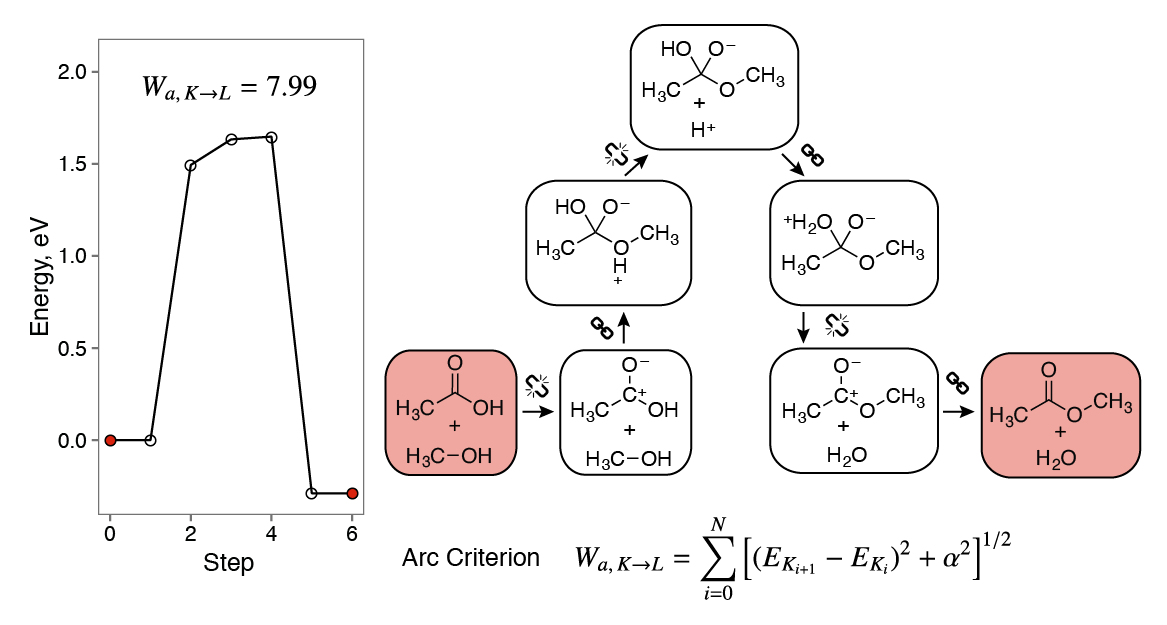
What are good chemical heuristics? For polar reactions, breaking bonds into ionic fragments and making bonds from ions are a sensible starting point. The heuristics-aided quantum chemistry (HAQC)3 method uses bond-breaking and bond-making heuristics to construct models of polar organic reactions, complete with the corresponding energy profiles. With the help of additional kinetic heuristics, we are able to distinguish feasible reaction pathways from infeasible ones. The arc heuristic criterion3 accounts for changes in energy along the reaction pathway while penalizing long reaction sequences.
- (a) Schlegel, H. B. Geometry optimization. WIREs Comput. Mol. Sci. , 1, 790. (b) Henkelman, G.; Jóhannesson, G.; Jónsson, H. Methods for finding saddle points and minimum energy paths. In Theoretical methods in condensed phase chemistry; Schwartz, S. D., Ed.; Kluwer: Dordrecht, ; Vol. 5, pp. 269–302.
- Pearl, J. Heuristics: Intelligent search strategies for computer problem solving. Addison-Wesley: Reading, MA, .
- Rappoport, D.; Galvin, C. J.; Zubarev, D. Yu.; Aspuru-Guzik, A. Complex chemical reaction networks from heuristics-aided quantum chemistry. J. Chem. Theory Comput., , 10, 897–907.
Networked Chemistry
Networked Chemistry
Once considered as little more than an academic curiosity, complex reaction networks are now found everywhere—in metabolism, in origins of life, and in many more reaction flasks than previously realized.1 These reaction networks are composed of many parallel and competing reaction pathways, branching and merging in a complex pattern with tens or even hundreds of chemical species involved.
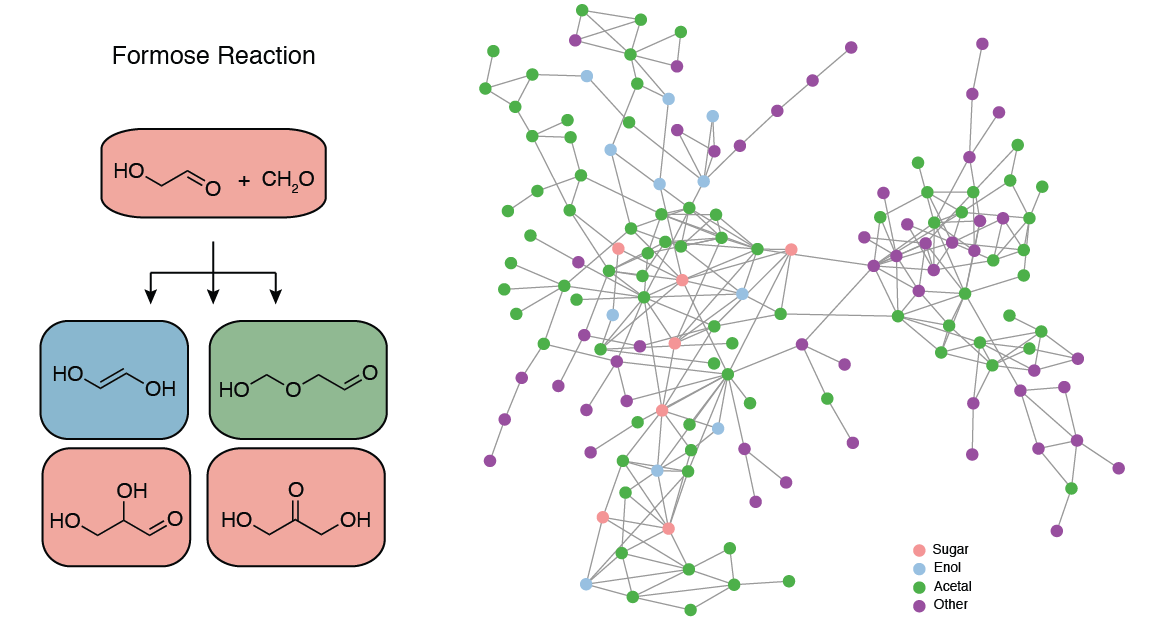
Perhaps the longest-known complex reaction network, the formose reaction, was obtained by Alexander Butlerov in 1861 by treating an aqueous formaldehyde solution with base. The result is a complex mixture of sugars, sugar alcohols, and other products, with > 50 individual substances identified thus far.2 What's more, the formose reaction starts out slow and then suddenly accelerates, a property known as autocatalysis. Because the formose reaction produces sugars—compounds central to cell metabolism—in an autocatalytic manner, it is often thought of as a key component in the origins of life on Earth.
The above model of the formose reaction network constructed by reacting glycolaldehyde (HOCH2CH=O) with two molecules of formaldehyde (CH2=O) by means of the heuristics-aided quantum chemistry (HAQC)3 approach has 149 nodes and 445 edges and includes all triose and tetrose sugars, among many other compounds. Moreover, the aldotetrose molecule is shown to undergo retroaldol cleavage into two glycolaldehyde molecules, thus closing the autocatalytic loop and demonstrating the standout feature of the formose reaction.
- Jeong, H.; Tombor, B.; Oltvai, Z. N.; Barabási, A.-L. The large-scale organization of metabolic networks. Nature, , 407, 651–654.
- (a) Boutlerow, A. Formation synthétique d'une substance sucrée. C. R. Acad. Sci. Paris, , 53, 145–147. (b) Breslow, R. On the mechanism of the formose reaction. Tetrahedron Lett., , 1, 22–26.
- Rappoport, D.; Galvin, C. J.; Zubarev, D. Yu.; Aspuru-Guzik, A. Complex chemical reaction networks from heuristics-aided quantum chemistry. J. Chem. Theory Comput., , 10, 897–907.
Conducting Molecules
Conducting Molecules
The promise of molecular electronics lies in the precise control, with which chemists can create molecular structures. New experimental results in molecular electronics challenge our understanding of conductance phenomena in materials. Molecules that do not carry electric current by the traditional drift mechanism, such as oligopeptides, can act as tunneling conductors.1
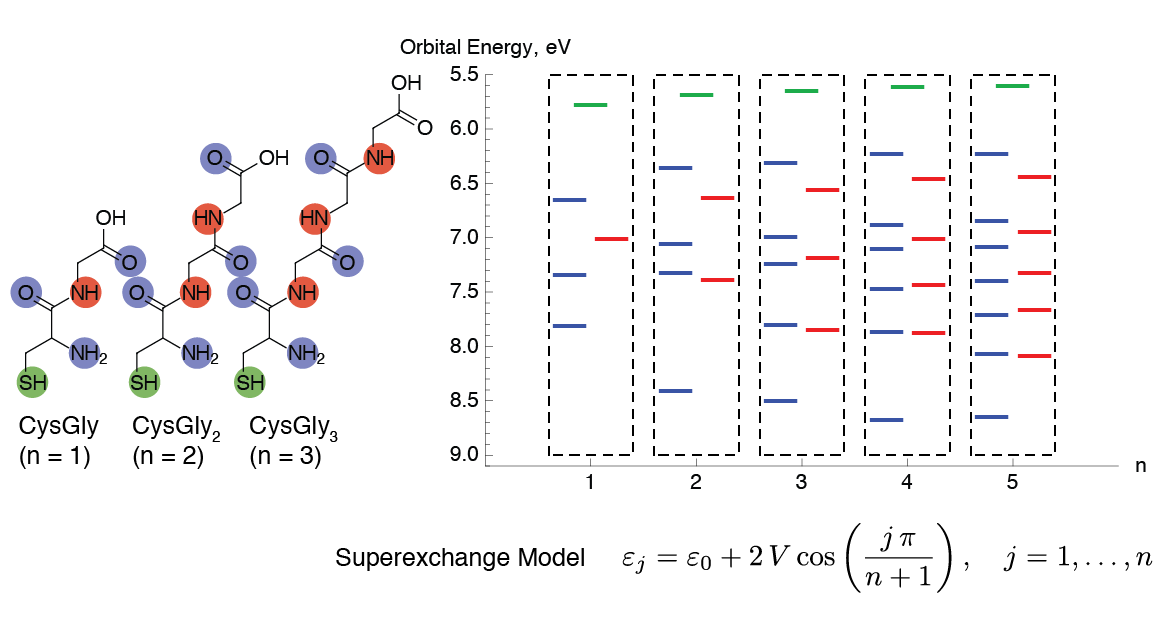
A simple model for understanding the tunneling conductance through molecules is the superexchange model.2 It assumes that the molecule has available a series of localized orbitals interacting with each other. Molecules with stronger interactions between neighboring groups are expected to be better tunneling conductors than molecules with weaker interactions.
For linear molecules, the superexchange model predicts a characteristic splitting pattern in the orbital energies. Using the computed values of the in-plane and out-of-plane peptide bond orbitals from density functional theory (DFT) calculations, we are able to derive the key parameters V and ε0 of the superexchange model for oligopeptides CysGlyn (n=1, …, 5).3
- Jortner, J.; Nitzan, A.; Ratner, M. A. Foundations of molecular electronics—Charge transport in molecular conduction junctions. In Introducing molecular electronics; Cuniberti, G., Richter, K., Fagas, G., Eds.; Springer: Berlin, ; pp. 13–54.
- Kosloff, R.; Ratner, M. A. Superexchange-assisted through-bridge electron transfer: Electronic and dynamical aspects. Isr. J. Chem., , 30, 45–48.
- Baghbanzadeh, M.; Bowers, C. M.; Rappoport, D.; Żaba, T.; Gonidec, M.; Al-Sayah, M. H.; Cyganik, P.; Whitesides, G. M. Charge tunneling along short oligoglycine chains. Angew. Chem. Int. Ed., , 54, 14743–14747.
Encoding 3D Molecular Structure
Encoding 3D Molecular Structure
The crucial first step toward a deeper understanding of chemical structure is the definition of a suitable non-empirical distance function expressing similarities between chemical and spatial structures of molecules. In conformational studies of small molecules and proteins, root-mean-square deviation (RMSD) has been the tool of choice for quantifying structural similarity. Unfortunately, RMSD cannot be applied to molecules of different composition.
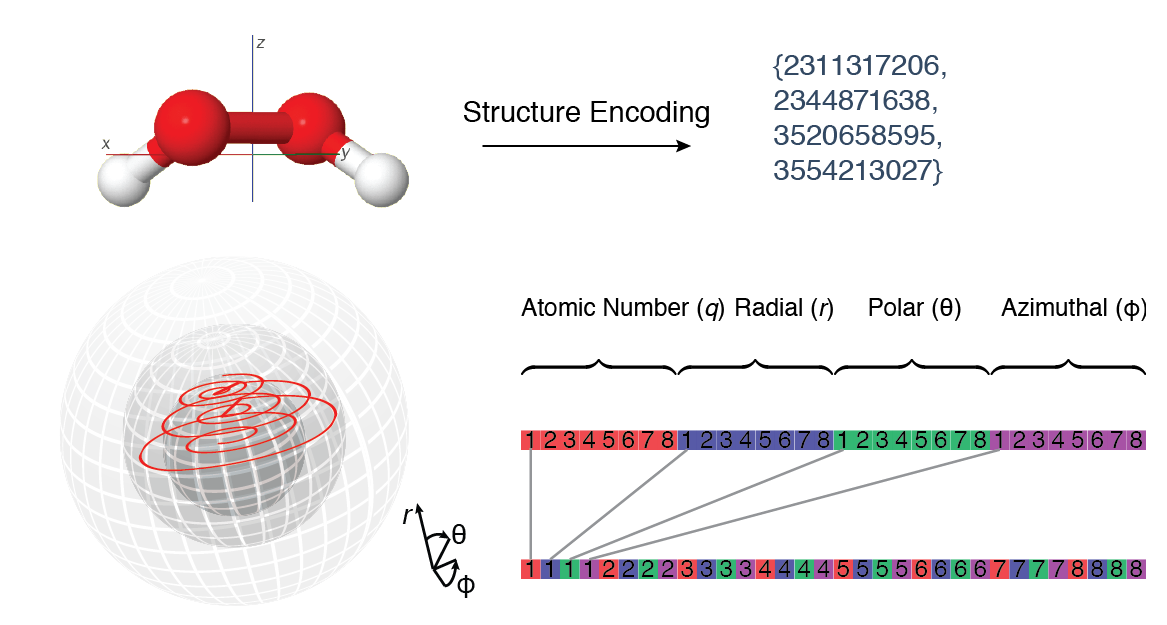
Our starting point is the 3D molecular structure. The atomic coordinates are first converted to a translationally and rotationally invariant representation. A discrete spherical grid is constructed with the center of mass as the origin. The nuclear charge q along with the discretized spherical polar coordinates r, θ, and ϕ form a four-dimensional discrete metric space. In order to preserve locality, the positions of the four-dimensional space (q, r, θ, and ϕ) are encoding usign a space-filling curve, e.g., Morton's Z-curve.1
Thanks to the discretization and traversal along a space-filling curve], the atomic and spatial properties are encoded as a fixed-length binary string, while the distances in the Cartesian space are approximated by string distances (Levenshtein distance is an example) or set distances (Hausdorff distance). Unlike RMSD and similar properties, string and set distances are well-defined for objects of different cardinality, i.e., molecules of different size or composition2.
- (a) Sagan, H. Space-filling curves. Springer: New York, NY . (b) Morton, G. M. A computer-oriented geodetic data base; and a new technique in file sequencing; Technical Report for IBM Ltd.: Ottawa, Canada, .
- Jasrasaria, D.; Pyzer-Knapp, E. O.; Rappoport, D.; Aspuru-Guzik, A. Space-filling curves as a novel crystal structure representation for machine learning models. , arXiv:1608.05747.
Journals and Book Chapters
- Rappoport, D.; Furche, F. Photoinduced intramolecular charge transfer in 4-(dimethyl)-aminobenzonitrile—A theoretical perspective. J. Am. Chem. Soc. , 126, 1277–1284.
- Bark, T.; Zelewsky, von, A.; Rappoport, D.; Neuburger, M.; Schaffner, S.; Lacour, J.; Jodry, J. Synthesis and stereochemical properties of chiral square complexes of iron(II). Chem. Eur. J. , 10, 4839–4845.
- Rappoport, D.; Furche, F. Analytical time-dependent density functional derivative methods within the RI–J approximation, an approach to excited states of large molecules. J. Chem. Phys. , 122, 064105.
- Furche, F.; Rappoport, D. Density functional methods for excited states: Equilibrium structure and electronic spectra. In Computational Photochemistry; Olivucci, M., Ed.; Elsevier: Amsterdam, , pp. 93-128.
- Rappoport, D.; Furche, F. Excited States and Photochemistry. In Time-Dependent Density Functional Theory; Marques, M. A. L.; Ullrich, C. A.; Nogueira, F.; Rubio, A.; Burke, K.; Gross, E. K. U., Eds.; Springer: Berlin, Heidelberg, , pp. 337–354.
- Rappoport, D.; Furche, F. Lagrangian approach to molecular vibrational Raman intensities using time-dependent hybrid density functional theory. J. Chem. Phys. , 126, 201104.
- Chow, H. S.; Constable, E. C.; Frantz, R.; Housecroft, C. E.; Lacour, J.; Neuburger, M.; Rappoport, D.; Schaffner, S. Conformationally-locked metallomacrocycles—Prototypes for a novel type of axial chirality. New J. Chem. , 33, 376–385.
- Rappoport, D., Furche, F. Structure of Endohedral Fullerene Eu@C74. Phys. Chem. Chem. Phys. , 11, 6353–6358.
- Rappoport, D.; Crawford, N. R. M.; Furche, F.; Burke, K. Approximate Density Functionals: Which Should I Choose? In Encyclopedia of Inorganic Chemistry. Computational Inorganic and Bioinorganic Chemistry; Solomon, E. I.; King, R. B.; Scott, R. A., Eds.; Wiley: Chichester, , pp. 159–172.
- Saikin, S. K.; Olivares-Amaya, R.; Rappoport, D.; Stopa, M.; Aspuru-Guzik, A. On the chemical bonding effects in the Raman response: Benzenethiol adsorbed on silver clusters. Phys. Chem. Chem. Phys. , 11, 9401–9411.
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. , 133, 134105.
- Saikin, S. K.; Chu, Y.; Rappoport, D.; Crozier, K. B.; Aspuru-Guzik, A. Separation of Electromagnetic and Chemical Contributions to Surface-Enhanced Raman Spectra on Nanoengineered Plasmonic Substrates. J. Phys. Chem. Lett. , 1, 2740–2746.
- Rappoport, D.; Shim, S.; Aspuru-Guzik, A. Simplified Sum-Over-States Approach for Predicting Resonance Raman Spectra. Application to Nucleic Acid Bases. J. Phys. Chem. Lett. , 2, 1254–1260.
- Parkhill, J. A.; Rappoport, D.; Aspuru-Guzik, A. Modeling Coherent Anti-Stokes Raman Scattering with Time-Dependent Density Functional Theory: Vacuum and Surface Enhancement. J. Phys. Chem. Lett. , 2, 1849–1854.
- Rappoport, D. Basis-set quality and basis-set bias in molecular property calculations. ChemPhysChem, , 12, 3404–3413.
- Watson, M. A.; Rappoport, D.; Lee, E. M. Y.; Olivares-Amaya, R.; Aspuru-Guzik, A. Electronic structure calculations in arbitrary electrostatic environments. J. Chem. Phys. , 136, 024101.
- Rappoport, D.; Hutter, J. Excited-State Properties and Dynamics. In Fundamentals of Time-Dependent Density Functional Theory; Marques, M. A. L.; Maitra, N. T.; Nogueira, F. M. S.; Gross, E. K. U.; Rubio, A., Eds.; Springer: Berlin Heidelberg, , pp. 317–336.
- Olivares-Amaya, R.; Rappoport, D.; Munoz, P. A.; Peng, P.; Mazur, E.; Aspuru-Guzik, A. Can Mixed-Metal Surfaces Provide an Additional Enhancement to SERS? J. Phys. Chem. C, , 116, 15568–15575.
- Widom, J. R.; Rappoport, D.; Perdomo-Ortiz, A.; Thomsen, H.; Johnson, N. P.; Hippel, von, P. H.; Aspuru-Guzik, A.; Marcus, A. H. Electronic transition moments of 6-methyl isoxanthopterin—A fluorescent analogue of the nucleic acid base guanine. Nucleic Acids Res., , 41, 995–1004.
- Widom, J. R.; Lee, W.; Perdomo-Ortiz, A.; Rappoport, D.; Molinski, T. F.; Aspuru-Guzik, A.; Marcus, A. H. Temperature-dependent conformations of a membrane supported zinc porphyrin tweezer by 2D fluorescence spectroscopy. J. Phys. Chem. A, , 117, 6171–6184.
- Lockett, M. R.; Lange, H.; Breiten, B.; Héroux, A.; Sherman, W.; Rappoport, D.; Yau, P. O.; Snyder, P. W.; Whitesides, G. M. The binding of benzoarylsulfonamide ligands to human carbonic anhydrase is insensitive to formal fluorination of the ligand. Angew. Chem. Int. Ed., , 52, 7714–7717.
- Rappoport, D.; Galvin, C. J.; Zubarev, D. Yu.; Aspuru-Guzik, A. Complex chemical reaction networks from heuristics-aided quantum chemistry. J. Chem. Theory Comput., , 10, 897–907.
- Bowers, C. M.; Liao, K.-C.; Yoon, H. J.; Rappoport, D.; Baghbanzadeh, M.; Simeone, F. C; Whitesides, G. M. Introducing ionic and/or hydrogen bonds into the SAM//Ga2O3 top-interface of AgTS/S(CH2)nT//Ga2O3/EGaIn junctions. Nano Lett., , 14, 3521–3526.
- Cabalo, J. B.; Saikin, S. K.; Emmons, E. D.; Rappoport, D; Aspuru-Guzik, A. A state-by-state investigation of destructive interference in resonance Raman spectra of neutral tyrosine and tyrosinate anion with the simplified sum-over-states approach. J. Phys. Chem. A, , 118, 9675–9686.
- Jinich, A.; Rappoport, D.; Dunn, I.; Sanchez-Lengeling, B.; Olivares-Amaya, R.; Noor, E.; Bar Even; A.; Aspuru-Guzik, A. Quantum chemical approach to estimating the thermodynamics of metabolic reactions. Sci. Rep., , 4, 7022.
- Hellweg, A.; Rappoport, D. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. Phys. Chem. Chem. Phys, , 17, 1010–1017.
- Bowers, C. M.; Liao, K.-C.; Żaba, T.; Rappoport, D.; Breiten, B.; Baghbanzadeh, M.; Krzykawska, A.; Cyganik, P.; Whitesides, G. M. Characterizing the metal–SAM interface in tunneling junctions. ACS Nano, , 9, 1471–1477.
- Zubarev, D. Yu.; Rappoport, D.; Aspuru-Guzik, A. Uncertainty of prebiotic scenarios: The case of the non-enzymatic reverse tricarboxylic acid cycle. Sci. Rep., , 5, 8009.
- Baghbanzadeh, M.; Bowers, C. M.; Rappoport, D.; Żaba, T.; Gonidec, M.; Al-Sayah, M. H.; Cyganik, P.; Whitesides, G. M. Charge tunneling along short oligoglycine chains. Angew. Chem. Int. Ed., , 54, 14743–14747.
- Bowers, C. M.; Rappoport, D.; Baghbanzadeh, M.; Simeone, F. C.; Liao, K.-C.; Semenov, S. N.; Żaba, T.; Cyganik, P.; Aspuru-Guzik, A.; Whitesides, G. M. Tunneling across SAMs containing oligophenyl groups. J. Phys. Chem. C, , 120, 11331–11337.
- Markovich, T.; Blood-Forsythe, M.; Rappoport, D.; Kim, D.; Aspuru-Guzik, A. Calibration of the many-body dispersion range-separation parameter. , arXiv:1605.04987.
- Jasrasaria, D.; Pyzer-Knapp, E. O.; Rappoport, D.; Aspuru-Guzik, A. Space-filling curves as a novel crystal structure representation for machine learning models. , arXiv:1608.05747.
- Baghbanzadeh, M.; Bowers, C. M.; Rappoport, D.; Zaba, T.; Yuan, L.; Kang, K.; Liao, K.-C.; Gonidec, M.; Rothemund, P.; Cyganik, P.; Aspuru-Guzik, A.; Whitesides, G. M. Anomalously Rapid Tunneling: Charge Transport across SAMs of Oligoethylene Glycol. J. Am. Chem. Soc. , 139, 7624–7631.
- Parker, S. M.; Rappoport, D.; Furche, F. Quadratic Response Properties from TDDFT: Trials and Tribulations. J. Chem. Theory Comput. , 14, 807–819.
- Sawaya, N.; Rappoport, D.; Tabor, D. P.; Aspuru-Guzik, A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano , 21, 6410–6420.
- Cafferty, B. J.; Yuan L.; Baghbanzadeh, M.; Rappoport, D.; Beyzavi, M. H.; Whitesides; G. M. Charge Transport through Self‐Assembled Monolayers of Monoterpenoids. Angew. Chem. Int. Ed. , 58, 8097–8102.
- Rappoport, D. Reaction Networks and the Metric Structure of Chemical Space(s) J. Phys. Chem. A , 123, 2610–2620.
- Rappoport, D.; Aspuru-Guzik, A. Predicting Feasible Organic Reaction Pathways Using Heuristically Aided Quantum Chemistry J. Chem. Theory Comput. , 15, 4099–4112.
Scientific Presentations
- Practical aspects of molecular TDDFT calculations. 2nd International Workshop on TDDFT, , Benasque (Spain).
- Computational aspects of molecular TDDFT calculations. University of Illinois, , Urbana-Champaign, IL.
- Optimized Gaussian basis sets for linear and nonlinear response properties. 44th Symposium on Theoretical Chemistry, , Ramsau (Austria).
- TDDFT in chemistry and biochemistry III and IV. TDDFT Winter School, , Benasque (Spain).
- New approaches to analysis of resonant Raman spectra. 240th ACS National Meeting, , Boston, MA.
- Optimized basis sets and accuracy of electronic excitation energies in molecules. 242th ACS National Meeting, , Denver, CO.
- Prediction of coherent anti-stokes Raman (CARS) and surface-enhanced CARS spectra using density functional theory. 242th ACS National Meeting, , Denver, CO.
- Parametrized exciton dynamics in multichromophoric aggregates. 48th Symposium on Theoretical Chemistry, , Karlsruhe (Germany).
- New theoretical approaches to resonant and nonlinear Raman spectroscopies. Edgewood Chemical Biological Center, , Edgewood, MD.
- Construction and properties of prebiotic chemical reaction networks. 248th ACS National Meeting, , San Francisco, CA.
- Reaction networks. A simplification attempt. Advanced Methods for De Novo Prediction Of Chemical Reaction Networks Workshop, , Telluride, CO.
- Exploring reaction mechanisms with heuristics-aided quantum chemistry. 254th ACS National Meeting, , Washington, DC.